

Bonnes pratiques de pharmacovigilance Module VI : collecte et soumission des ICSR en 2026
Temps de lecture : 11 min
Points clés à retenir
- GVP Module VI : il encadre la collecte, la gestion et la soumission des cas individuels de sécurité (ICSR) pour tous les médicaments à usage humain dans l’Espace économique européen.
- Délais stricts : 15 jours calendaires pour les réactions graves, 90 jours pour les non-graves, sous peine de sanctions réglementaires.
- Addendum I et II : deux textes essentiels (gestion des doublons et masquage des données personnelles) qui ont renforcé la qualité et la confidentialité des données en 2025-2026.
- Outils numériques : EudraVigilance, EVWEB et les API sont désormais la norme ; la formation du personnel reste un facteur clé de conformité.
Qu’est-ce que le Module VI des GVP ?
Sur le terrain, on constate que le Module VI des bonnes pratiques de pharmacovigilance (GVP) est souvent perçu comme le « cœur opérationnel » du système européen de pharmacovigilance. Il détaille les procédures de collecte, de gestion, d’évaluation et de soumission des cas individuels de sécurité (ICSR). Publié dans sa révision 2, enrichi de deux addenda (I sur la gestion des doublons, II sur le masquage des données), il constitue la référence réglementaire pour tout titulaire d’autorisation de mise sur le marché (TAMM) et toute autorité compétente.
Pour être précis : ce module s’applique aux médicaments à usage humain, y compris les vaccins, les produits biologiques, les médicaments en accès compassionnel, et même les études cliniques post-AMM. Il ne faut pas le confondre avec d’autres modules : le Module VII traite des rapports périodiques de sécurité (PSUR), le Module IX de la gestion des signaux. Le Module VI, lui, est celui du quotidien – celui de la notification « brute ».
Petite astuce de labo : dans ma pratique, je distingue toujours le Module VI du VII en disant « le VI, c’est l’instantané ; le VII, c’est le bilan annuel ». Cette analogie aide les équipes à se repérer.
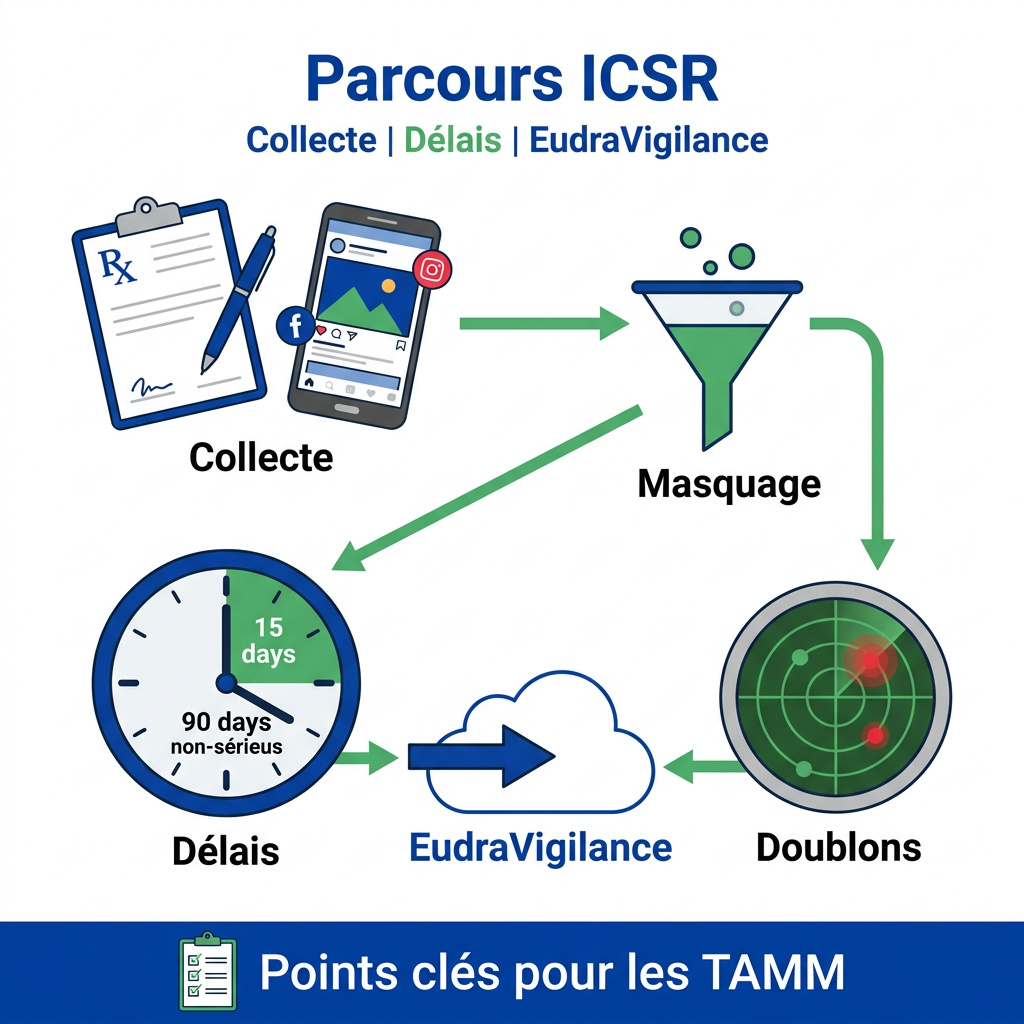
Pourquoi le Module VI est-il crucial en 2026 ?
En avril 2026, la pharmacovigilance européenne connaît une transformation numérique accélérée. En 2025, ce sont plus de 2 millions d’ICSR qui ont été soumis via EudraVigilance, dont environ 500 000 via la plateforme EVWEB. Le digital a bouleversé la collecte : médias sociaux, applications mobiles, forums santé – autant de sources désormais surveillées (section VI.B.1.1.4).
C’est une question qu’on me pose souvent : « Ces nouvelles sources ne génèrent-elles pas plus de doublons ou de données incomplètes ? » Oui, et c’est exactement pour cela que l’Addendum I sur la détection des doublons est devenu indispensable. Avant sa mise en œuvre, le taux de doublons atteignait 20 % dans certaines bases. Après application des algorithmes renforcés, ce taux a diminué d’environ 30 %.
Mon conseil : intégrer les API de soumission directe depuis les outils de vos partenaires (hôpitaux, cliniques) réduit les saisies manuelles et améliore la qualité des données.
Définitions clés selon le Module VI
Il est essentiel de maîtriser le vocabulaire, car une erreur de libellé peut entraîner un délai de soumission inapproprié. Voici les notions fondamentales :
- Événement indésirable (EI) : tout événement nocif non intentionnel, qu’il soit lié ou non au médicament. Exemple : un patient se plaint de nausées après la prise d’un antibiotique – c’est un EI.
- Réaction indésirable grave (RIG) : événement qui entraîne la mort, une mise en danger, une hospitalisation, une incapacité persistante, une anomalie congénitale ou une situation médicalement significative. Elle doit être notifiée dans les 15 jours.
- ICSR : dossier complet comprenant au moins un patient, un médicament, une réaction et un rapporteur.
- Source primaire : professionnel de santé, patient, ou tout autre individu qui rapporte l’événement.
Dans la pratique quotidienne, je vérifie toujours le statut de gravité dès la réception du premier appel : un simple coup d’œil aux critères (décès, hospitalisation…) évite des erreurs de chronométrage.
Processus de collecte des ICSR
La collecte est le maillon le plus « humain » de la chaîne. Voici les sources obligatoires :
- Professionnels de santé : médecins, pharmaciens, infirmiers – ils doivent transmettre aux TAMM ou aux autorités.
- Patients : depuis les campagnes de sensibilisation, les notifications directes augmentent.
- Études cliniques post-AMM : les investigateurs rapportent les événements.
- Littérature médicale : les TAMM doivent surveiller les publications.
- Internet et médias sociaux : une section spécifique (VI.B.1.1.4) impose une recherche active sur les forums et les réseaux identifiés.
Sur le terrain, on constate que la collecte via les réseaux sociaux est sous-utilisée. Attention à ne pas négliger cette source : l’EMA attend une preuve de surveillance. Un délai de 3 jours ouvrables est accordé entre la réception du signalement par le TAMM et l’enregistrement dans la base de données. Pour les cas graves, ce délai est réduit à 24 heures pour la déclaration initiale.
Gestion et qualité des données
Une fois collecté, le cas doit être validé. Un ICSR est considéré complet s’il contient au moins un patient identifiable, un médicament suspecté, une réaction et un rapporteur. Ensuite, deux étapes critiques :
Gestion des doublons (Addendum I)
L’Addendum I, entré en vigueur fin 2025, impose aux TAMM de mettre en place un algorithme de détection des doublons basé sur au moins trois des critères suivants : nom du patient, âge, sexe, médicament, réaction, date. Avant cet addendum, le taux de doublons était estimé à 20 %. Depuis son application, une réduction d’environ 30 % a été observée. Mon conseil : utilisez un logiciel de pharmacovigilance qui intègre ces règles automatiques ; le travail manuel devient vite ingérable au-delà de quelques milliers de cas.
Masquage des données personnelles (Addendum II)
L’Addendum II, en vigueur depuis avril 2026, renforce la protection des données. Tout ICSR soumis à EudraVigilance doit être pseudonymisé : les noms, adresses, photos, pièces d’identité sont masqués de manière irréversible. C’est une obligation dans le cadre du RGPD et de la transparence des données de santé. « Petit » détail technique : les formats acceptés sont le XML ICH E2B(R3), et tout fichier mal formaté est rejeté par le portail.
Soumission des ICSR à EudraVigilance
La soumission est l’étape finale. Les délais réglementaires sont impératifs :
- Réactions graves : 15 jours calendaires à compter de la réception par le TAMM.
- Réactions non graves : 90 jours calendaires.
Les modalités de soumission sont multiples : EVWEB (interface web pour les petits volumes), FTP (pour les soumissions par lots), ou API (pour l’automatisation). En 2025, 500 000 rapports ont été soumis via EVWEB. Un TAMM moyen traite entre 10 000 et 50 000 cas par an. Le délai de traitement cible pour les cas graves est de 72 heures après la validation initiale.
Attention à : les études cliniques post-AMM sont soumises aux mêmes délais. Beaucoup d’équipes oublient que le jour de la connaissance de l’événement par l’investigateur est le point de départ, pas la date de réception du rapport écrit.
Pièges fréquents et bonnes pratiques
Fort de mon expérience de formatrice, je recense les trois erreurs les plus courantes :
- Codage MedDRA incohérent : environ 15 % des soumissions comportent au moins une incohérence (ex : terme trop général, mauvaise version du dictionnaire).
- Non-respect des délais pour les études cliniques : parce que l’événement est recueilli dans un cadre de recherche, certains pensent qu’il n’est pas soumis aux délais GVP. C’est faux.
- Manque de formation du personnel : les techniciens et les pharmacovigilants changent, les procédures évoluent. Il faut des audits internes réguliers.
Petite astuce de labo : mettez en place un fichier de suivi des cas avec des alertes automatiques à J-10 pour les cas graves, J-60 pour les non-graves. Cela vous laisse une marge de sécurité.
Évolutions à venir (2026-2027)
La pharmacovigilance ne cesse d’évoluer. Voici ce qui se profile :
- Harmonisation internationale : ICH E2B(R3) devient le standard mondial, et des extensions pour les vaccins et les produits biologiques sont en préparation.
- Intelligence artificielle : l’EMA a lancé des projets pilotes utilisant l’IA pour détecter les doublons et coder automatiquement les réactions. Les premiers résultats montrent une réduction de 40 % du temps de gestion des cas.
- Produits biologiques et biosimilaires : les obligations de traçabilité (nom de marque + numéro de lot) deviennent plus strictes pour surveiller les réactions spécifiques.
Mon conseil : suivez les publications de l’EMA sur ces pilotes et préparez vos équipes à intégrer ces outils dès leur déploiement. L’avenir de la pharmacovigilance est dans l’automatisation intelligente, mais la vérification humaine reste indispensable.
Conclusion
Le Module VI des bonnes pratiques de pharmacovigilance est bien plus qu’un texte réglementaire : c’est un guide opérationnel pour garantir la sécurité de millions de patients européens. Pour résumer : collectez rigoureusement, validez rapidement, masquez les données personnelles, détectez les doublons, soumettez dans les délais, et formez vos équipes. La mise en œuvre de l’Addendum I et II en 2025-2026 a élevé les standards de qualité, et les outils numériques (EudraVigilance, EVWEB, API) sont vos meilleurs alliés.
Pour aller plus loin, je vous invite à consulter la documentation officielle de l’EMA mise à jour en avril 2026, accessible sur leur site.
Questions fréquemment posées
Qu’est-ce que le module VI des bonnes pratiques de pharmacovigilance (GVP) ?
C’est le cadre réglementaire européen qui définit les procédures de collecte, de gestion et de soumission des cas individuels de sécurité (ICSR) pour tous les médicaments à usage humain.
Quels sont les délais de soumission des ICSR selon le module VI ?
15 jours calendaires pour les réactions graves, 90 jours pour les non-graves, à compter de la réception de l’information par le TAMM.
Comment gérer les doublons de cas individuels de sécurité (Addendum I) ?
En mettant en place un algorithme basé sur au moins trois critères (nom, âge, sexe, médicament, réaction, date) et en vérifiant manuellement les cas suspects.
Qu’est-ce que le masquage des données personnelles dans EudraVigilance (Addendum II) ?
La pseudonymisation des ICSR avant soumission : suppression des identifiants directs (nom, adresse, photos) pour respecter le RGPD tout en permettant l’analyse médicale.
Quelles sources de collecte sont obligatoires pour les ICSR ?
Les professionnels de santé, les patients, les études cliniques post-AMM, la littérature médicale, et les médias sociaux (selon section VI.B.1.1.4).
Quelle est la différence entre le module VI et le module VII (PSUR) ?
Le module VI traite des cas individuels (ICSR) en temps réel, tandis que le module VII concerne les rapports périodiques de sécurité (PSUR) – des bilans d’ensemble.
Comment soumettre un ICSR via EVWEB ?
En accédant au portail de l’EMA avec des identifiants de sécurité, en remplissant le formulaire XML ICH E2B(R3) et en le transmettant par l’interface web.
Quels sont les critères de gravité d’un événement indésirable selon le module VI ?
Décès, mise en danger vitale, hospitalisation ou prolongation, incapacité persistante, anomalie congénitale, ou tout autre événement médicalement significatif.
Le module VI s’applique-t-il aux médicaments en essai clinique ?
Non, les essais cliniques sont régis par le Règlement (UE) 536/2014. Le module VI s’applique aux médicaments déjà autorisés (post-AMM), y compris en études post-autorisation.
Quelles sont les conséquences d’un non-respect des délais de soumission ?
Des sanctions réglementaires pouvant aller jusqu’à la suspension de l’autorisation de mise sur le marché pour le TAMM, en plus d’un impact sur la sécurité des patients.

Pharmacienne biologiste & Rédactrice scientifique
Pharmacienne biologiste diplômée depuis 15 ans, j’ai exercé en laboratoire d’analyses médicales privé avant de me tourner vers la rédaction scientifique et la formation professionnelle. Spécialisée dans la vulgarisation des pratiques de laboratoire, j’accompagne aujourd’hui les professionnels de santé et les étudiants à travers des contenus clairs et documentés.
Expertises : Biologie médicale • Biotechnologies • Matériel de laboratoire • Réglementation ISO • Formation continue


